RESEARCH
◥
These design principles have proven useful for
fast, reliable catalyst activation and are widely
used for new catalyst discovery.
REPORT
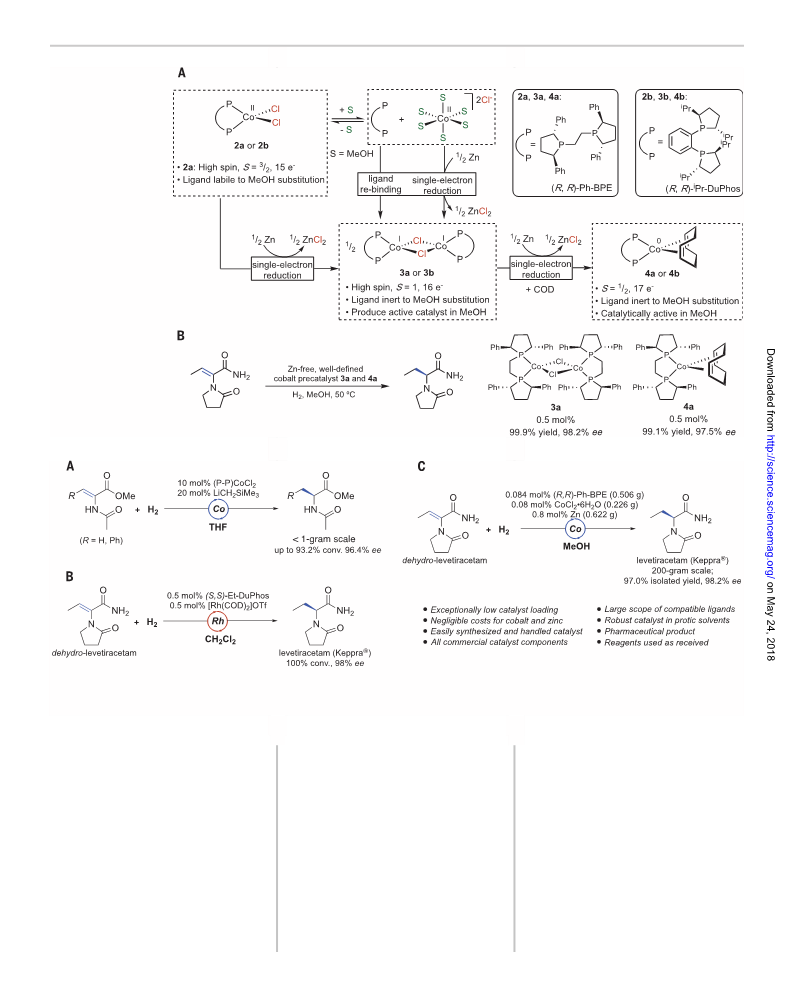
Our laboratory has reported that two-carbon-
bridged, C2-symmetric (bis)phosphines support
highly active and enantioselective cobalt cata-
lysts for the asymmetric hydrogenation of simple
dehydro-a–amino acid derivatives (6). Although
these catalysts are state-of-the-art among first-
row metals and provide an important demon-
stration of the promise of earth-abundant metals
in asymmetric alkene hydrogenation, major lim-
itations include the use of pyrophoric activators
such as LiCH2SiMe3, extreme air sensitivity of
the catalyst, and lack of reactivity among many
classes of phosphines, likely owing to catalyst
deactivation by irreversible loss of ligand (Fig. 1E).
Isolated organometallic compounds such as (R,R)-
QuinoxP*Co(CH2SiMe3)2 and (R,R)-iPr-DuPhosCo
(CH2SiMe3)2 (iPr, isopropyl), albeit more active,
require multistep organometallic syntheses and
special handling that are likely impractical for
industrial application. Here we describe advances
in cobalt-catalyzed asymmetric alkene hydrogena-
tion, where mechanistic insights into ligand dis-
sociation equilibria and the unique properties of
the first-row transition metals are leveraged to
address fundamental limitations of existing ca-
talysts. Two sequential single-electron reductions
of substitutionally labile Co(II) complexes result
in formation of more robust catalysts in situ. This
advance in catalyst activation enabled the dis-
covery of scores of effective metal-ligand combina-
tions for asymmetric hydrogenation, culminating
in the use of low catalyst loadings for the prac-
tical, pilot-scale synthesis of an API.
The hydrogenation of dehydro-levetiracetam
(1) was selected for initial catalyst development
studies to highlight the challenges associated
with API synthesis. The corresponding chiral
product, levetiracetam (Keppra), is a widely used
medication for epilepsy (16). In one patented
route (17), levetiracetam was prepared by as-
ymmetric hydrogenation using an optimized
condition of 0.5 mole % (mol %) of in situ–
generated [(S,S)-Et-DuPhosRh(COD)][OTf] in di-
chloromethane (Et, ethyl; COD, 1,5-cyclooctadiene;
OTf, triflate). The relatively high catalyst loading
and use of a noncoordinating, chlorinated sol-
vent reflects the challenges associated with hydro-
genation of 1 as a poorly coordinating substrate
(17) with limited conformational flexibility for
achieving two-point binding (18).
CATALYSIS
Cobalt-catalyzed asymmetric
hydrogenation of enamides enabled
by single-electron reduction
Max R. Friedfeld,1 Hongyu Zhong,1 Rebecca T. Ruck,2
Michael Shevlin,2* Paul J. Chirik1*
Identifying catalyst activation modes that exploit one-electron chemistry and overcome
associated deactivation pathways will be transformative for developing first-row transition
metal catalysts with performance equal or, ideally, superior to precious metals. Here we
describe a zinc-activation method compatible with high-throughput reaction discovery that
identified scores of cobalt-phosphine combinations for the asymmetric hydrogenation of
functionalized alkenes. An optimized catalyst prepared from (R,R)-Ph-BPE {Ph-BPE, 1,2-
bis[(2R,5R)-2,5-diphenylphospholano]ethane} and cobalt chloride [CoCl2·6H2O] exhibited
high activity and enantioselectivity in protic media and enabled the asymmetric synthesis
of the epilepsy medication levetiracetam at 200-gram scale with 0.08 mole % catalyst
loading. Stoichiometric studies established that the cobalt (II) catalyst precursor
(R,R)-Ph-BPECoCl2 underwent ligand displacement by methanol, and zinc promoted facile
one-electron reduction to cobalt (I), which more stably bound the phosphine.
symmetric catalysis with soluble metal
complexes has transformed the preparation
of single enantiomers in the pharmaceuti-
cal, fragrance, and fine-chemical industries
(1, 2). Because different antipodes of chiral
states separated by one electron, often to the
detriment of catalytic chemistry (5). Although
considerable progress has been made (6, 7), state-
of-the-art catalysts with iron, cobalt, and nickel
lack many of the favorable properties associated
with precious metal catalysts that facilitate scale
up. Alkene hydrogenation catalysts with earth-
abundant metals are typically air- and moisture-
sensitive, requiring rigorously dried solvents; are
intolerant of many polar functional groups found
in APIs; and have insufficient activity to be ap-
plied industrially.
Asymmetric hydrogenation and other enantio-
selective metal-catalyzed reactions often rely on
the successful relay of stereochemical information
from a chiral ligand to the substrate (8). There-
fore, understanding and controlling ligand co-
ordination and dissociation equilibria are key
to enabling catalyst stability and communicating
stereochemical information. Unlike other tactics
for improving catalyst performance, rational con-
trol of catalyst activation and deactivation are
challenging and often circumvented by increased
catalyst loadings (9–11). With classic transition
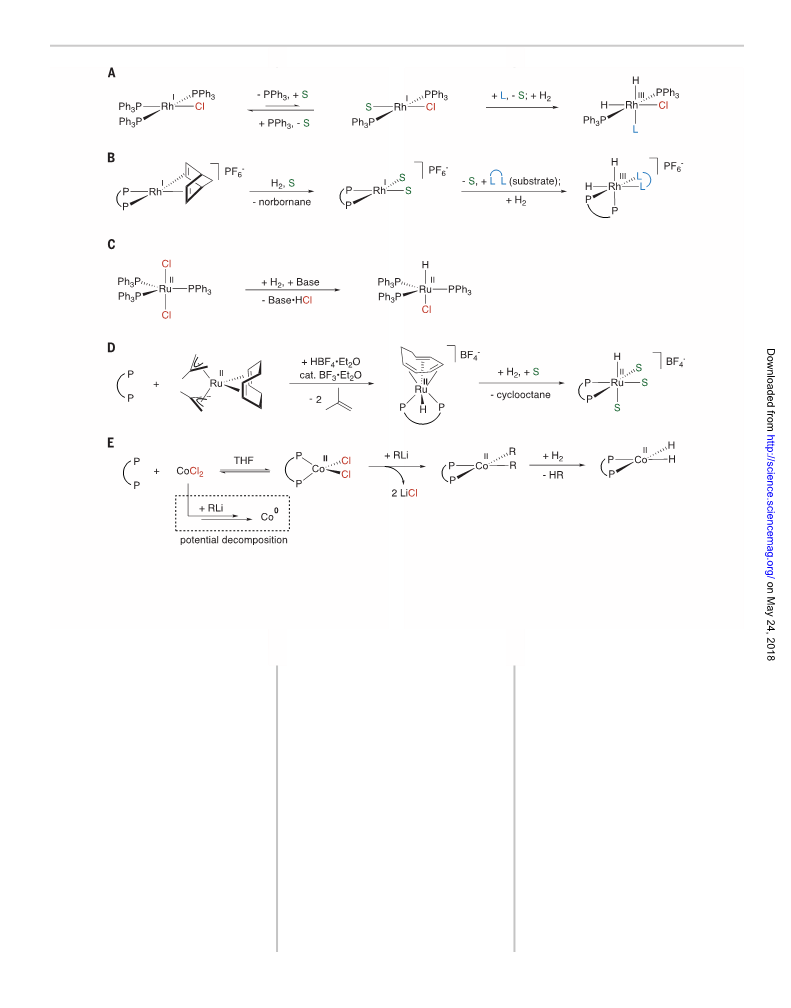
metal catalysts such as Wilkinson’s (Ph3P)3RhCl
(12) and (Ph3P)3RuCl2 (13) (Ph, phenyl), catalyst
activation and, ultimately, performance is limited
by phosphine dissociation equilibria and halide
coordination (Fig. 1, A and C). These limitations are
overcome with weakly coordinating anions and
hydrogenation of ancillary diene or triene ligands
to create open coordination sites (Fig. 1, B and
D), as exemplified by the Schrock-Osborn–type
catalysts [(P,P)Rh(diene)][X] (14), where X is
any noncoordinating anion, and the cationic
ruthenium catalyst [(P-P)Ru(H)(triene)][BF4] (15).
A
molecules often exhibit distinct biological prop-
erties, the U.S. Food and Drug Administration
has strict requirements for single-enantiomer
drugs, and the importance of asymmetric trans-
formations in the pharmaceutical industry will
continue to grow. Beginning with Knowles’s syn-
thesis of the Parkinson’s medication L-dopa by
rhodium-catalyzed asymmetric alkene hydro-
genation (3), catalysis by homogeneous catalysts
containing precious metals with tunable lig-
ands has revolutionized the approach to single-
enantiomer active pharmaceutical ingredients
(APIs).
The widespread application of asymmetric
hydrogenation, particularly in the pharmaceutical
industry, has motivated efforts to identify cata-
lysts based on earth-abundant first-row transi-
tion metals rather than traditionally used precious
metals (4). In alkene hydrogenation catalysis,
rhodium and iridium catalysts operate by pre-
dictable, two-electron cycles involving oxidative
addition and reductive elimination [for example,
M(I)-M(III)]. However, compared to their heavier
congeners, first-row transition metals have kinet-
ically and thermodynamically accessible oxidation
Initial studies on hydrogenation of 1 relied on
high-throughput experimentation to evaluate re-
action variables, including solvents, cobalt sources,
activators, temperature, and catalyst loadings
(tables S1 to S6). A remarkable solvent dependence
was identified: Protic solvents such as methanol
(MeOH), ethanol, and trifluoroethanol provided
the highest yields and enantiomeric excesses
(tables S1 to S4). These unexpected results in-
dicated that cobalt hydrides competent for en-
antioselective alkene hydrogenation could, despite
their anticipated hydricity, be formed in protic
solvents. The use of alcohol solvents has been
prevalent since the discovery of enantioselective
1Department of Chemistry, Princeton University, Princeton, NJ
08544, USA. 2Department of Process Research and
Development, Merck Research Laboratories, Rahway, NJ 07065,
USA.
*Corresponding author. Email: michael_shevlin@merck.com
(M.S.); pchirik@princeton.edu (P.J.C.)
Friedfeld et al., Science 360, 888–893 (2018)
25 May 2018
1 of 5

 Friedfeld, Max R.
Friedfeld, Max R.